eg technology writes:
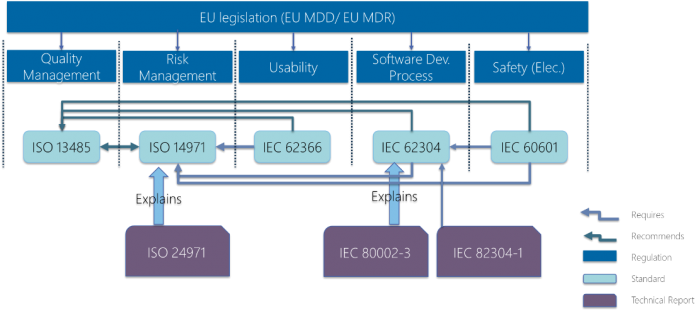
ISO 14971 is required by both IEC 62304 – the standard for medical device software development, IEC 60601 - covering basic safety and essential performance of medical electrical equipment and IEC 62366 – the application of usability engineering which has come under greater scrutinty with the EU MDR regulation.
Like many standards, ISO 14971 is written in a way that superficially makes sense but is not terribly helpful in giving practical advice on how to implement it. Although the companion document ISO/TR 24971 is very helpful and has some practical suggestions.
When to start your risk management
Once you have an idea for a device and some idea of how you would build it (you have done some proof of principle work or have some experimental data), you start your medical device development.
It is very difficult to retrofit compliance with 14971 and so it’s important to get started early. The first document you need is a plan. 24971 contains good guidance on what the plan should cover (see section 4.4) and it should not be too onerous to put this together. As part of this, you need to set out the level of risk that you are prepared to accept. 24971 contains examples severity levels (5.5.5 table 2 or table 4) probability levels (5.5.5 table 3 and table 5).
![]()
These levels should lead to a matrix. Each combination of severity and probability should result in a risk being deemed 'Acceptable', 'Not Acceptable' or 'Borderline'.
We would always recommend a medical device development programme should be broken into phases and each phase should contain the core steps of:
Risk Analysis – What are the hazardous situations and the potential risks?
Risk Evaluation – what is the probability that a risk will occur and the severity of its impact?
Risk Control – what can you do to control or mitigate the risk?
Throughout the process it’s important to remember you aren’t aiming for zero risk, but at the end of your development you will need to evaluate the residual risk and justify why the benefit of your device outweighs the risk.
How to start the risk analysis?
![]()
We would suggest that the first thing to do is characterise the device. 24971 Annex A has helpful questionnaire that you should think about and provide Yes/No answers to depending on whether they apply to your device. For those that do apply, the questions also act as prompts for a more in-depth consideration of the most important (risky) aspects of your device.
This should help you to create a list of hazards that are present in your device. There is a fairly exhaustive list in Annex C of 14971. IEC 62304 also suggests some software related hazards.
We would then suggest considering potential Harms. These are much more specific to each device but should generally include what the “not working” harm is – i.e. what is the harm if the user cannot use the device. The FDA Maude database is publicly accessible and contains reported incidents associated with medical devices. If your device is similar to or competes with a more established ones, you may get some insight into harms by seeing what has been reported for them.
We would suggest that you score the severity of the harm at this stage. This should involve clinical input to assess the real-world impact of the harm occurring.
Having established a list of Hazards and Harms, we tend to do a Preliminary Hazard Analysis. This is to identify potential issues at an early stage, before getting into detailed design. To make this work, remember that it is preliminary and therefore don’t go too detailed or too exhaustive early. You are looking for the main risks present in the system and how you can mitigate them. There will be time to be more detailed via an FMEA at a later stage.
For each hazard, this involves answering questions like:
How might people, things or the environment come into contact with the hazard?
What could fail that could cause a hazardous situation?
How could each of the harms come to pass?
When constructing this list, you also need to think about reasonably foreseeable misuse. This term is widely misunderstood. Foreseeable misuse covers
Users making mistakes
Users not following or ignoring instructions
It should not cover users behaving maliciously or deliberately attempting to cause harm. You then need to evaluate those risks and score them for both probability and severity using the tables from your risk management plan. For any risks deemed 'Not Acceptable' or 'Borderline', you need to document risk control measures, reducing them to the point that you can make that final determination that the benefits outweigh the risks.
No further work is required on any risks that are acceptable.
How are you going to construct the evidence chain?
When finally submitting your device for approval, you will need to demonstrate that:
You have carried out your risk management plan
You have identified and documented risks
You have created requirements that are part of your risk control measures
You have tested and met those requirements
To pass your audit/inspection, you will need to be able demonstrate a chain from a hazardous sequence of events to a risk to a requirement to a test and to a test result.
It is worth getting your strategy for this in place early because retrofitting it is much harder.
We use a system called MatrixALM for the chain of requirements, tests and test results. So, we document the link from a risk control to a requirement to achieve this.
Summing up
We find that considering hazards and harms prior to attempting the preliminary hazard analysis (PHA) or failure mode effect analysis (FMEA) helps bound the issues being considered.
The traceability from risk to requirement to test and verification is key to achieving compliance.
Our experience has convinced us that taking the requirements of 14971 into consideration early on in a medical device development programme will make the product better and make achieving certification easier.
For more information or to chat with one of the eg technology team about your product design and development requirements, please contact us:
via email on design@egtechnology.co.uk
by giving us a call on +44 01223 813184
or by clicking here